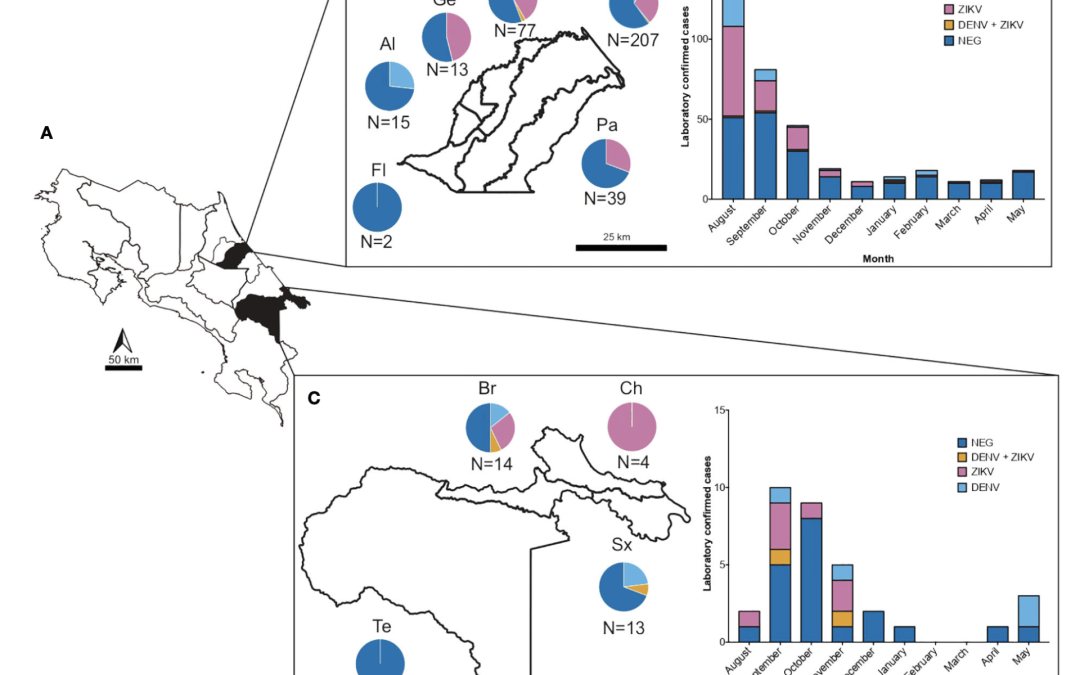
The increase in incidence and geographical expansion of viruses transmitted by the Aedes mosquitoes, such as dengue (DENV) and zika (ZIKV) in the Americas, represents a burden for healthcare systems in tropical and subtropical regions. These and other under-detected arboviruses co-circulate in Costa Rica, adding additional complexity to their management due to their shared epidemiological behavior and similarity of symptoms in early stages. Since diagnostics of febrile illness is mostly based on clinical symptoms alone, we gathered acute-phase serum and urine from 399 samples of acute dengue-like cases from two healthcare facilities of Costa Rica, during an outbreak of arboviruses from July 2017 to May 2018, and tested them using molecular and serological methods. The analyses showed that of the clinically presumptive arbovirus cases that were reported, only 39.4% (n=153) of the samples were confirmed positive by RT-PCR to be DENV (DENV (10.3%), CHIKV (0.2%), ZIKV (27.3%), or mixed infections (1.5%). RT-PCR for other alphaviruses and flaviviruses, and PCR for Leptospira sp were negative. Furthermore, to assess flavivirus positivity in post-acute patients, the negative sera were tested against Dengue-IgM. 20% of sera were found positive, confounding even more the definitive number of cases, and emphasizing the need of several distinct diagnostic tools for accurate diagnostics. Molecular characterization of the prM and E genes from isolated viruses revealed that the American/Asian genotype of DENV-2 and the Asian lineage of ZIKV were circulating during this outbreak. Two different clades of DENV-2 American/Asian genotype were identified to co-circulate in the same region and a difference in the platelet and leukocyte count was noted between people infected with each clade, suggesting a putative distinct virulence. Our study sheds light on the necessity for healthcare strategies in managing arbovirus outbreaks, emphasizing the importance of comprehensive molecular and serological diagnostic approaches, as well as molecular characterization. This approach aids in enhancing our understanding of the clinical and epidemiological aspects of arboviral diseases during outbreaks. Our research highlights the need to strengthen training programs for health professionals and the need to increase research-based on laboratory evidence for diagnostic accuracy, guidance, development and implementation of public health interventions and epidemiological surveillance.