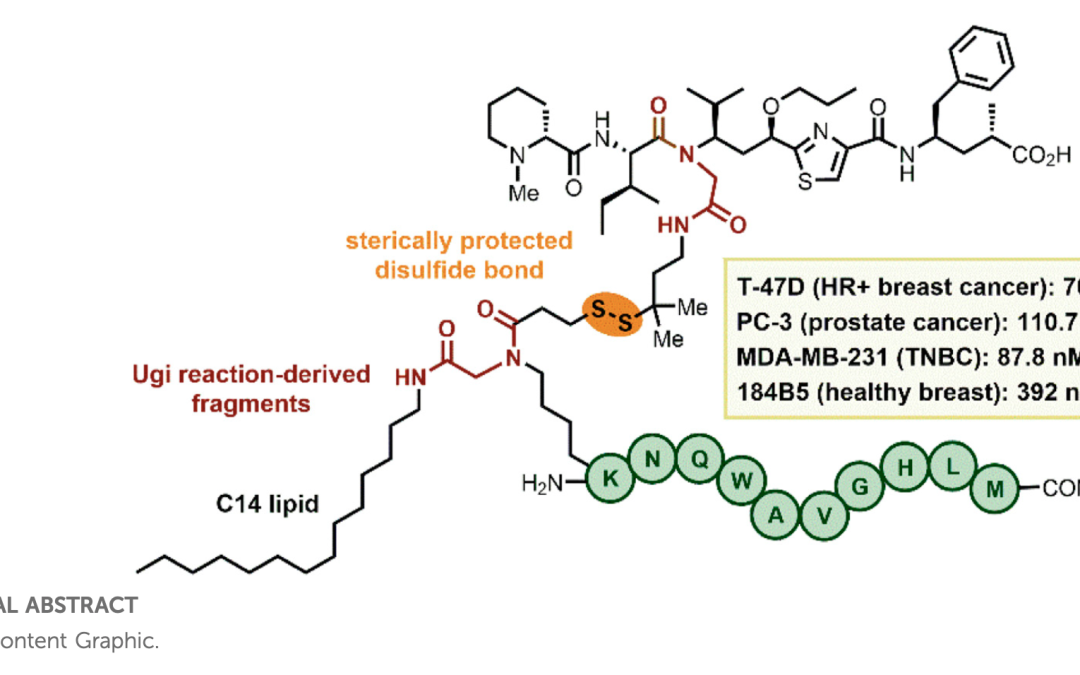
Exploration of the Tertiary Amide Chemical Space of Dolastatin 15 Analogs Reveals New Insights into the Structure–Anticancer Activity Relationship
Dolastatins are a class of naturally occurring antimitotic peptides that have inspired the development of some of the most active and widely used anticancer agents. Here, we report on the development of synthetic methodologies for the preparation of parallel libraries of small peptides inspired by dolastatin 15 and itsanalogs cemadotin and tasidotin. The approaches rely on the use of either one or multiple Ugi-multicomponent reactions to generate amide N-substituted dolastatin-like skeletons, which allow the exploration of tertiary amide chemical spaces that have not been assessed previously. Evaluation of the anticancer activity in a variety of cancer cells showed that introducing a tertiary amide at the C-terminal fragment or by replacement of a proline residue does not lead to an increment in the anti-proliferative activity. The microtubule-disrupting capacity of the new dolastatin analogs was studied and compared with other potent antimitotic agents, thereby shedding light on mechanistic details of their anti-proliferative activity.